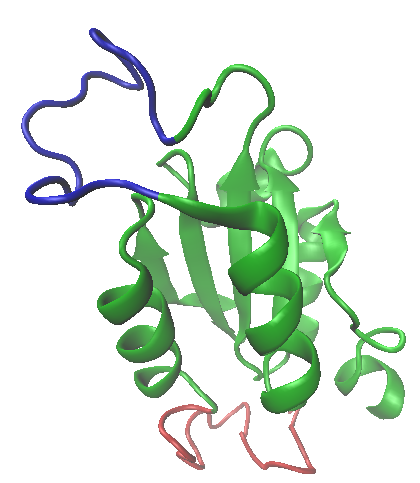
This month I have picked a protein I first heard about from a crystallographer, visiting Sheffield around 1981. What interested me was that the structure had been refined to a very high resolution (less than one angstrom, in the early 1980s!). What also interested me was that this was a protein with no known function. So what was the point of all that effort! The molecule itself is pretty unremarkable (see the image left). It contains two alpha helices oriented into a Y-shape, linked by a short, constrained loop. The N-and C-termini seem to have considerable freedom, with a small amount of beta sheet, but surprisingly perhaps, the protein crystallises very easily.

The molecular envelope shown right, shows crambin to be a globular molecule with a well defined shape. Without any knowledge of its function, the surface of the long helix is presented for interaction and the N and C termini could re-fold around a small ligand or another macromolecule. But there is no evidence of a metal ion or any significant space for a small ligand or substrate. After all, it contains less than 50 amino acids, which makes it an ideal candidate for NMR spectroscopy.
 The protein was initially isolated from an Abyssinian cabbage (or kale) and is now known to belong to the family of toxins called thionins. [The source of proteins used by biochemists would make a nice Blog post for the future!] The key to the stability of the terminal segments is (as the name thionin suggests) the disulphide bond. The sequence of crambin is shown below, with the Cys residues highlighted. If you look at the representation shown left, you can see the yellow sulphurs and the small network of disulphide bonds that contribute to the stability of the structure. This is a feature of many extracellular proteins, including immunoglobulins. The analysis of structures at such high resolution provides a molecular framework for defining the precise geometry of such bonding phenomena and provide a nice
The protein was initially isolated from an Abyssinian cabbage (or kale) and is now known to belong to the family of toxins called thionins. [The source of proteins used by biochemists would make a nice Blog post for the future!] The key to the stability of the terminal segments is (as the name thionin suggests) the disulphide bond. The sequence of crambin is shown below, with the Cys residues highlighted. If you look at the representation shown left, you can see the yellow sulphurs and the small network of disulphide bonds that contribute to the stability of the structure. This is a feature of many extracellular proteins, including immunoglobulins. The analysis of structures at such high resolution provides a molecular framework for defining the precise geometry of such bonding phenomena and provide a nice
TTCCPSIVARSNFNVCRLPGTPEALCATYTGCIIIPGATCPGDYAN
experimental opportunity to address the role of disulphides in stabilisation and in protein folding pathways of proteins in general. Try mapping the bonds from the structure onto the sequence. You will immediately appreciate that primary structures must be considered in three dimensions in order to fully appreciate the significance of sequence conservation!
The possible application of this plant product in cancer treatment is being investigated, but remains at an early stage to date. One other point I would like to draw your attention to is a comparison between methods of structure determination. X-ray crystallography "prefers" proteins of several hundred or more amino acids in each polypeptide, whereas Nuclear Magnetic Resonance (NMR) spectroscopy "prefers" proteins with molecular weights below 20 000 (<200 amino acids). These rules aren't hard and fast, but they do significantly improve the probability of obtaining a high resolution data set (required to fix the position of side-chain atoms). The structural representation on the right was obtained by NMR. NMR structure determination generates an "ensemble" of structures that are consistent with the spectral data. (See here for an introduction to protein NMR). The first thing you realise is that some parts of the protein are better defined than others. In X-ray crystallography, any significant "flexibility" in a protein structure usually prevents the assignment of electron density in that region and this may mean that this section of a protein is not included in the deposited structural file (it is usually pointed out in the publication). In the case of crambin, the NMR structure suggests that the N and C termini are pretty rigid. A consequence of the disulphide bonding, explaining why the protein is so compact and probably explains why it such an amenable molecule for obtaining high resolution atomic data.
So finally, why has so much effort been invested by structural biologists in a molecule of such poorly defined function? This is an important issue in Science in general: what should we (as tax payers, versus say drug companies) spend our money on? Molecules like crambin can help establish the fundamental principles of protein structure. Some medically important molecules may be difficult to purify, may be unstable or may yield poor diffraction (in the case of X ray crystallography) or may be difficult to solubilise and show poor spectral resolution (for NMR). The insight we gain from "well-behaved" proteins can help us fill in the gaps with molecules that we can easily recognise as being of societal value. Moreover, workhorses like crambin can help us push the envelope of techniques like X-ray crystallography and NMR, which may then make it more likely that we can interpret the data from proteins that are less well-behaved! One final point is that structure determination alone can rarely determine the function of a molecule. It may be that we need to solve the structures of the entire proteome of humans before we are ready to comprehensively link structure and function in Biology!

 There are some words you will need to be familiar with in understanding this post. Diurnal is derived from the Latin meaning of the day or daily and comes from the word dyeu to shine (diamond?). Nocturnal, I think you will know refers to the night and finally, circadian (combined here with rhythm), comes from circa (about or around) and day and is an adjective that describes a process that occurs on a 24 hour cycle. Some species are awake in the day and sleep at night, like us (and I will come back to the problems that shift work and long plane journeys can cause) and others are nocturnal, like bats: sleeping all day and foraging at night. I hope you can see here how evolutionary adaptation is linked to the motion of the planets. Let's look at what they discovered, before I return to the planets!
There are some words you will need to be familiar with in understanding this post. Diurnal is derived from the Latin meaning of the day or daily and comes from the word dyeu to shine (diamond?). Nocturnal, I think you will know refers to the night and finally, circadian (combined here with rhythm), comes from circa (about or around) and day and is an adjective that describes a process that occurs on a 24 hour cycle. Some species are awake in the day and sleep at night, like us (and I will come back to the problems that shift work and long plane journeys can cause) and others are nocturnal, like bats: sleeping all day and foraging at night. I hope you can see here how evolutionary adaptation is linked to the motion of the planets. Let's look at what they discovered, before I return to the planets!
 Finally, beyond the fundamental importance of molecular basis of Circadian Rhythms, why may they be important in respect of our health and well-being? As I mentioned above, by flying in the face of our diurnal nature, the regulation of the PER system needs to adapt: this happened when you fly a long distance or you adopt the working habits of a badger and work shifts. Many people choose to work permanent nights, and must therefore re-configure their body clock. Try it once when you are not forced to! [I should mention that the PER network of regulation interacts with light sensors (cryptochromes) thereby providing the cell (and the body) with valuable cues which fine tune the body clock. It is also becoming clear that there is a correlation between the action of certain drugs and the body clock. If you are interested you can read these "rapid response" articles that appeared just after the announcement.
Finally, beyond the fundamental importance of molecular basis of Circadian Rhythms, why may they be important in respect of our health and well-being? As I mentioned above, by flying in the face of our diurnal nature, the regulation of the PER system needs to adapt: this happened when you fly a long distance or you adopt the working habits of a badger and work shifts. Many people choose to work permanent nights, and must therefore re-configure their body clock. Try it once when you are not forced to! [I should mention that the PER network of regulation interacts with light sensors (cryptochromes) thereby providing the cell (and the body) with valuable cues which fine tune the body clock. It is also becoming clear that there is a correlation between the action of certain drugs and the body clock. If you are interested you can read these "rapid response" articles that appeared just after the announcement.